0 1
0 2
0 3
0 4
Integrated Drug Discovery
Sygnature Discovery's collaborative teams drive efficiency in the drug development process.
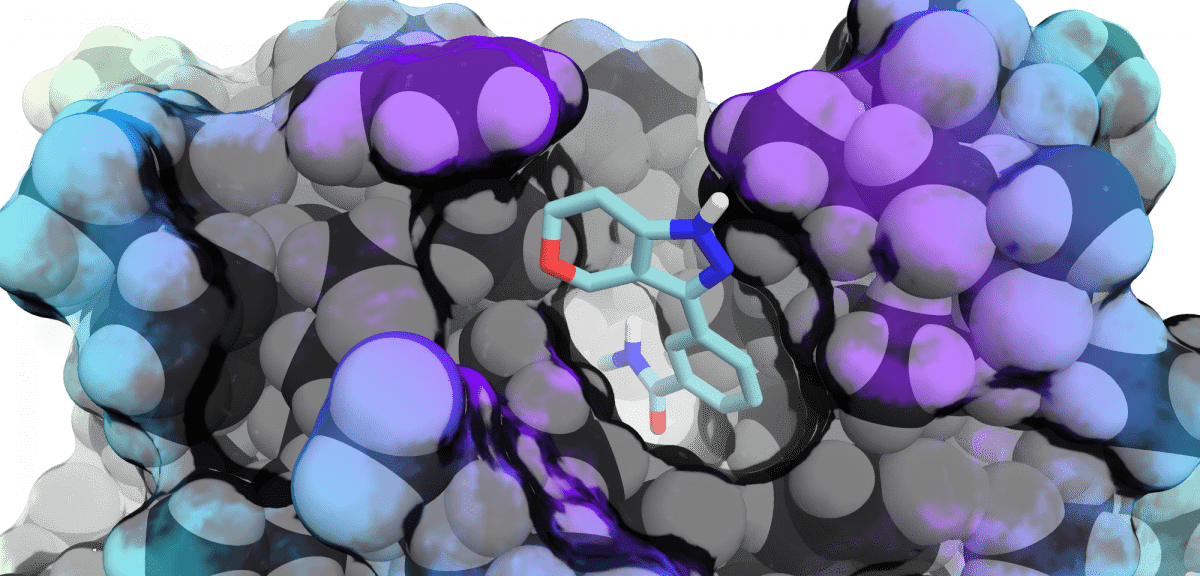
Discover moreComputational Chemistry
Our expert Computer Aided Drug Design (CADD) team can help accelerate your drug discovery journey to...
Discover moreIn Vivo Pharmacology
Discover our integrated in vivo pharmacology services, blending consultancy and experimental testing...
Discover moreForm and Formulation
Advanced Drugs Formulation Development Services: Partnering for Successful Preclinical Studies and C...
Discover moreBioscience
At Sygnature Discovery, our extensive bioscience knowledge and collaborative approach in drug discov...
Discover moreChemistry
Unlock Success in Small Molecule Drug Discovery with Sygnature Discovery's Medicinal Chemistry Team....
Discover moreDMPK Consultancy
Sygnature Discovery offers a range of DMPK consultancy services to drug discovery research teams wit...
Discover moreFibrotic Diseases
Sygnature Discovery’s integrated teams of medicinal chemists, bio scientists,...
Discover Fibrotic Diseases
Metabolic Diseases
Sygnature’s scientists have extensive experience in the field of...
Discover Metabolic Diseases
Oncology and
Immuno-oncology
We have successfully delivered more than 16 pre-clinical oncology...
Discover Oncology and
Anti Infectives
Sygnature have a history of enabling discovery success in...
Discover Anti InfectivesNeuroscience
Our scientists have a well-developed understanding of the challenges...
Discover Neuroscience
Inflammation and Immunology
Our scientists have a rich heritage in this therapeutic...
Discover Inflammation and Immunology